

Renormalization of Molecular Electronic Levels at Metal-Molecule Interfaces
Foundry users M. S. Hybertsen, G. W. Flynn, Columbia University
There is renewed interest in using organic molecules as components in nanoscale electronic and optoelectronic devices, and a critical need has emerged for improved understanding and control of optical and transport phenomena in organic molecular assemblies, particularly at the metal contact. A high concentration of such nanointerfaces is expected in organic-based optoelectronic devices, such as hybrid organic-inorganic nanoscale solar cells.
Frontier molecular electronic states, accessible experimentally via photoemission or transport measurements, dictate the nature of optical absorption, chemical reactivity, and transport at metal-organic interfaces. In this Foundry user project with Dr. Mark Hybertsen at Columbia University, first-principles theory was used to predict and understand the behavior of the electronic states responsible for conduction of simple aromatic molecules on a metal surface.
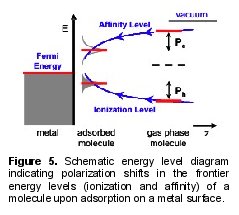
When a molecule is brought in contact with a metal, several physical effects will influence its ionization (highest occupied molecular orbital, HOMO) and affinity (lowest unoccupied molecular orbital, LUMO) levels. First, the self-consistent interaction between molecule and surface will rearrange the electron density and modify the alignment of frontier orbital energies. Second, electronic coupling to extended states in the metal will further shift orbital energies and broaden discrete molecular levels into resonances. Finally, the Coulomb interaction between the added hole (electron) associated with the ionization (affinity) level will result in a polarization of the metal substrate.
Calculated frontier orbital energy levels (heavy blue lines) with the indicated energy gap (red arrows) for benzene adsorbed flat on the graphite surface, plotted against the projected surface band structure of graphite: (a) DFT (LDA) energies, (b) GW quasiparticle energies, (c) GW quasiparticle energies of benzene in the gas phase. Inset in (a) shows a model of the adsorption geometry.
Our studies centered on level alignment at a prototypical metal-molecule contact, benzene physisorbed on graphite (0001), using the GW approximation for the electron self-energy, which explicitly accounts for many-electron interactions . We find that in the case where the molecule is weakly bound to the surface, the contacts strongly influence and alter the efficacy of the organic separate charge. The benzene HOMO-LUMO gap was computed to be 7.2 eV on graphite, substantially reduced from its calculated gas-phase value of 10.5 eV. (The local density approximation is found to be inadequate, predicting a gap of about 5 eV for the molecule in both environments.) Strong static polarization effects shift the benzene frontier orbitals, effectively narrowing the fundamental energy gap . Our study allowed us to develop a simple electrostatic model that can explain scanning-probe measurements of excited states of aromatic molecules physisorbed on the metal surfaces ,. This quantitative model should help to understand, identify, and design optimal metal-molecule contacts in organic optoelectronic devices, e.g. solar cells that will result in more efficient charge injection or collection.
Publication arising from this work: J.B. Neaton, M.S. Hybertsen, and S.G. Louie, "Renormalization of Molecular Electronic Levels at Metal-Molecule Interfaces," Physical Review Letters 97 (21), p.216405, (2006).
